-
![]()
![]()
Promotion im FG Materialmodellierung
28.06.2021
Dissertation von Frau Leonie Koch – Wir gratulieren!
Thema der Dissertation:„First-principles study of the defect chemistry and conductivity in sodium bismuth titanate“
-
![]()
![]()
Nanoidentation von Nanogläsern
16.04.2021
Einsichten aus atomistischen Computersimulationen des mechanischen Verhaltens
-
![]()
![]()
Semianalytischer Ansatz zur Beschreibung von Transportlücken in polykristallinem Graphen
16.04.2021
Einblicke in die Korngrenzeigenschaften von polykristallinem Graphen
-
![]()
![]()
Lösungshärtung von CrMnFeCoNi-basierten Hochentropielegierungen
16.04.2021
Zusammenarbeit der FG Physikalische Metallkunde und Materialmodellierung
-
![]()
![]()
Dislocation-toughened ceramics
16.04.2021
Zur Rolle der Versetzungsdichte in Strontiumtitanat
-
![]()
![]()
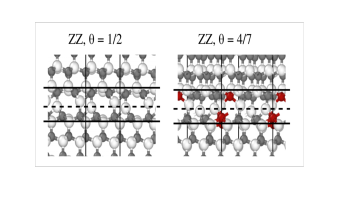
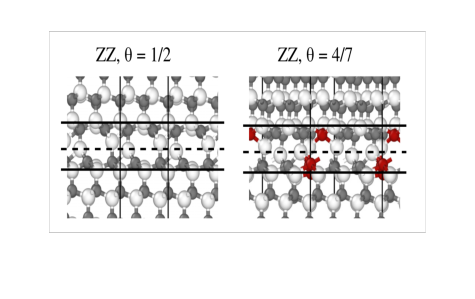
Kationenunordnung verbessert den superionischen Leiter NASICON
21.03.2021
Wie sich der Na-Transport im Festelektrolyten NASICON verbessern lässt
-
![]()
![]()
Materialmodellierung meets Kernphysik
12.03.2021
Vermessung und Simulation des elektromagnetischen Zerfall eines angeregten Lithium-Isotops mit beispielloser Präzision
-
![]()
![]()
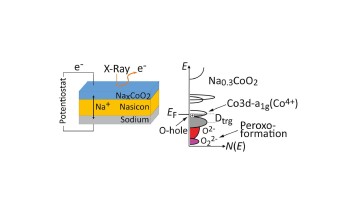
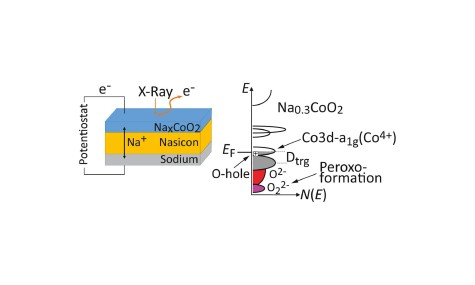
Ladungskompensation in Batteriekathoden aus Natriumkobaltat
09.02.2021
Elektronische Struktur einer Operando-Festkörperzelle mit Natriumkobaltatkathode
Zusammenspiel von Elelektronenspektrospie, Dünnschichtanalyse und Elektronenstrukturrechnungen
-
![]()
![]()
Inversionsdomänengrenzen in ZnO
04.02.2021
Aufklärung der elektronischen Struktur und Thermodynamik mit Hilfe von DFT Rechnungen
Wir haben die atomare und elektronische Struktur sowie die chemische Zusammensetzung von ZnO-Bikristallproben mit {0001} Inversionsdomänengrenzen (IDB) mit Hilfe von Berechnungen der Dichtefunktionaltheorie untersucht. Die Rechnungen zeigen, dass thermodynamisch bevorzugte IDBs durch vollständig (4-fach) koordinierte Atome gekennzeichnet sind und relativ geringe Grenzflächenenergien im Bereich von 45 bis 95 meV / Å^2 besitzen,l. Die elektronischen Eigenschaften des IDB weichen nur schwach von denen des volummenmaterials ab und sind gegenüber Druck- und Zugspannungen eher unempfindlich. Unsere Ergebnisse zeigen, dass experimentell beobachtete piezotronische Eigenschaften von Bikristallen keine intrinsische Eigenschaft des unberührten GB selbst sind, sondern Defekten, Verunreinigungen oder Dotierstoffen stammen. DIe hier identifizierten Niedrigenergiestrukturmodelle können auch auf andere IDBs vom wz- oder zb-Typ (z. B. GaN, AlN, SiC usw.) übertragbar sein.
-
![]()
![]()
Defektgetriebene flexochemische Phänomene in ferroischen Materialien
28.12.2020
Ein phänomenologischer theoretischer Ansatz
Dieser Übersichtsartikel konzentriert sich auf die phänomenologische Beschreibung des Einflusses elastischer Defekte auf die elektrophysikalischen Eigenschaften von nanoskaligen ferroelektrischer Materialien. Unter Verwendung des phänomenologischen Ansatzes von Landau-Ginzburg-Devonshire betrachten wir den Beitrag der elastischen Defekte (z. B. neutrale Sauerstoffleerstellen) zu den Phasenübergangstemperaturen, Phasendiagrammen, piezoresistiven, dielektrischen und polaren Eigenschaften dünner ferroelektrischer Filme und Nanopartikel. Besonderes Augenmerk legen wir auf die Rolle des flexoelektrischen Effekts, der Vegard-Dehnungen und -Spannungen, einschließlich ihrer Synergie, die als flexochemische Kopplung bezeichnet wird, sowie auf die defektbedingten Phänomene in Nanoferroics. Wir untersuchen den Einfluss der Migration mobiler geladener Defekte (z. B. Drift und Diffusion von Kationen oder Leerstellen von geladenem Sauerstoff) auf die Domänenstrukturentwicklung in dünnen ferroelektrischen Filmen. Wir diskutieren auch die Möglichkeiten der selbstorganisierten Ordnung ungeladener elastischer Defekte in dünnen verspannten Oxidfilmen und zeigen, dass eine thermodynamisch stabile ungeordnete Phase, räumlich modulierte Phasen und langreichweitig geordnete Phasen mit Defektschichten parallel oder senkrecht zur Substratebene möglich sind erscheinen im Phasendiagramm in Abhängigkeit von der Film-Substrat-Fehlanpassungsdehnung, der Konzentration elastischer Defekte und den Vegard-Koeffizienten. Da die langreichweitig geordneten ferroischen Phasen multiferroisch werden können, wenn einige elastische Dipole zu elektrischen werden, eröffnen die erhaltenen Ergebnisse den Weg zur Erzeugung und Steuerung fehlerhaft geordneter Überstrukturen durch Auswahl eines geeigneten Substrats und einer Defektkonzentration in dünnen ferroischen Filmen.
FG Materialmodellierung
Archiv
Archiv
















Kontakt
Prof. Dr. rer. nat. Karsten Albe
Fachgebietsleiter

albe@mm.tu-...
fax +49 6151 16-20965
Work
L6|01 211
Otto-Berndt-Str. 3
64206
Darmstadt
Gabriele Rühl
Teamassistenz

work +49 6151 16-21901
fax +49 6151 16-20965
Work
L6|01 210
Postfach 10 06 36
Otto-Berndt-Str. 3
64206
Darmstadt