-
![]()
![]()
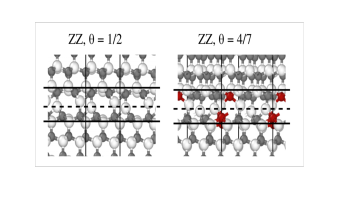
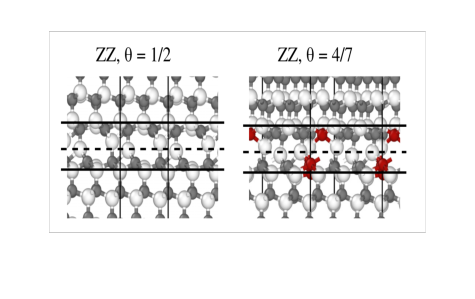
Interplay of cation order in the superionic conductor NASICON
2021/03/21
How to improve Na-transport in a solid electrolyte
-
![]()
![]()
Materials Modelling meets Nuclear Physics
2021/03/12
Measurement and simulation of the electromagnetic decay of an excited lithium isotope with unparalleled precision
-
![]()
![]()
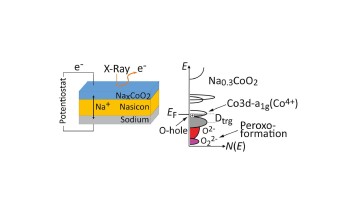
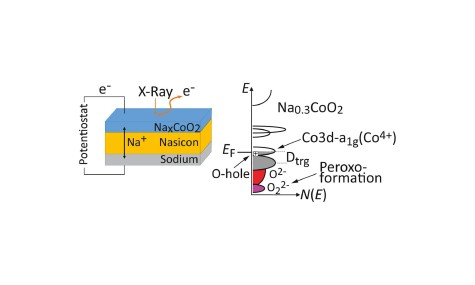
Charge compensation mechanism in sodium cobaltate electrode materials
2021/02/09
Electronic structure of an operando solid-state cell with sodium cobaltate cathode
Electron spectroscopy combined with thin film technology and electronic structure calculations
-
![]()
![]()
Inversion domain boundaries in ZnO
2021/02/04
Electronic structure and thermodynamics revealed by DFT calculations
We systematically investigated the atomic structure and chemical composition of ZnO bicrystal samples containing {0001} inversion domain boundaries (IDB) using density functional theory calculations. We find that thermodynamically favored IDBs are characterized by fully (4-fold) coordinated atoms and possess relatively low excess energies that range from 45 to 95meV/Å2, depending on the termination (Zn/Zn or O/O) and the exchange-correlation functional. The electronic properties of the GB deviate only weakly from those of the bulk and are rather insensitive towards compressive and tensile strains. Our results thus indicate that experimentally observed piezotronic properties of bicrystals are not an intrinsic property of the pristine GB itself, but originate, for example, from externally supplied trapped charges, defects, impurities, or dopants. Low-energy structure models identified here may also be transferable to other wz- or zb-type IDBs (e.g., GaN, AlN, SiC, etc.).
-
![]()
![]()
Defect driven flexo-chemical phenomena in ferroics
2020/12/28
A phenomenological theory
This review is focused on the phenomenological description of the elastic defects influence on the electrophysical properties of nanosized ferroics. Using the Landau-Ginzburg-Devonshire phenomenological approach we consider the contribution of the elastic defects (e.g. neutral oxygen vacancies) on the phase transitions temperatures, phase diagrams, piezoresistive, dielectric and polar properties of thin ferroelectric films and nanoparticles. We pay special attention to the role of flexoelectric effect, Vegard strains and stresses, including their synergy, called flexo-chemical coupling, on the defect-driven phenomena in nanoferroics. We explore the influence of mobile charged defects migration (e.g. the drift and diffusion of cations or charged oxygen vacancies) on the domain structure evolution in thin ferroelectric films. Also, we discuss the possibilities of the uncharged elastic defects self-ordering ordering in thin strained oxide films, and show that a thermodynamically stable disordered phase, spatially modulated phases, and long-range ordered phases with defect layers parallel or perpendicular to substrate plane can appear in the phase diagram in dependence on the film-substrate mismatch strain, concentration of elastic defects and Vegard coefficients. Since the long-range ordered ferroic phases can become multiferroic when some elastic dipoles become electric ones, the obtained results open the way to create and control defect-ordered superstructures by the choice of appropriate substrate and defect concentration in thin ferroic films.
-
![]()
![]()
Why superionic conductor can be optimized by thermal quenching
2020/12/28
Increase of Li ion conductivity in argyrodites by freezing anion site-disorder
In this joined work with W. Zeiers group (WWU Münster), we demonstrate synthetic control over the anion site-disorder in Li6 PS4Br. Site-disorder can be tailored by subjecting the material to different temperatures followed by quenching. Neutron powder diffraction and AIMD simulations show that changing site-disorder results in diferences in the local charges, which in turn directly affects the lithium distribution. A higher degree of site-disorder means more spatially distributed Li and shorter Li–Li distances responsible for the intercage jumps. The optimized site-disorder can subsequently lead to a fourfold increase of the ionic conductivity. This work provides a better understanding of how synthesis and post synthesis conditions affect the structure and the ionic transport in lithium argyrodites. This knowledge is a stepping-stone for engineering materials properties without changing the composition of the material itself.
-
![]()
![]()
Virtual Christmas Party
2020/12/19
MM X-Mas Extravaganza
Christmas party in virtual mode
-
![]()
![]()
And yet it moves
2020/11/27
LiNiO2, a dynamic Jahn−Teller System
In this joined work with researchers from BASF we studied the dynamics and cooperativity of JT distortions in LiNiO2. While static DFT calculations unequivocally advocate the stability of a monoclinic JT-distorted LNO, this is elusive to experiments: JT distortions are observed on a local scale, but the global crystallographic symmetry is systematically identified as a perfectly undistorted rhombohedral structure isostructural with LiCoO2. Thanks to molecular dynamics simulations, we monitored individual Ni−O bond lengths as a function of time and assessed that JT distortions indeed take place, but reorient dynamically. The statistical correlation analysis revealed that, although some extent of cooperativity might survive within one plane, correlation between different layers is for all practical purposesnonexistent. The continuous reorientation of the JT direction appears on average as though all Ni−O bonds were equal, whereas non-cooperativity between layers prevents the system from undergoing a macroscopic monoclinic shear; the combination of these two effects attributes to LNO the commonly accepted R3̅m symmetry. This result rationalizes the conundrum that, for nearly two decades, juxtaposed the clear JT activity of Ni3+ to the lack of macroscopic monoclinic shear. Quoting John Hasbrouck Van Vleck, who extended the JT theorem to ions in crystals, “it is a great merit of the JT effect that it disappears when not needed”. R3̅m is neither a local minimum nor a saddle point: it is the tip of the Mexican-hat-shaped potential, whereas the reorientation path goes along the warped trough around the maximum. Computational studies of LNO and directly related compounds should therefore avoid using R3̅m as the reference compound and use the true ground state (P21/c)instead; not only is using R3̅m conceptually wrong, but it also leads to non-negligible quantitative errors. R3̅m should still be used in the analysis of diffraction data on LNO.
-
![]()
![]()
Mechanical properties of glassy nanopillars
2020/11/27
A comparative, computational study of size effects in nanoglasses and homogeneous bulk glasses
We have investigated the influence of structure size on the mechanical properties of NG and HG nanopillars with 7 nm grain size and diameters ranging from 4.5 up to 54 nm by means of MD simulations. Simulations were done for two different glasses, namely, Cu 64 Zr 36 and Pd 80 Si 20 , as representatives of metal–metal and metal–metalloid systems, respectively. Different from previous studies, the NGs were produced by consolidation rather than Voronoi tessellation and thus have a more realistic microstructure. Our results show a clear difference in the deformation mode between NG and HG for the 36 and 54 nm nanopillars, independent of the glass type. While HG nanopillars exhibit a stress drop and strain localization developing in a shear band, NG nanopillars show ductile deformation behavior with softening at larger engineering strains and deformation by necking. The tensile ductility of about 13–15% found in our simulations is in agreement with 15% plastic strain observed for a 400 nm Sc 75 Fe 25 NG nanopillar using in situ tensile tests in a transmission electron microscope (Wang et al., 2015). HG and NG nanopillars with D = 4.5 nm, where the pillar diameter is smaller than the average grain size d = 7 nm of the NG, deform by necking since the nucleation of STZs on the surface is dominating. In the HG nanopillars with D = 9 and 18 nm, shear banding is more obvious in Cu 64 Zr 36 than in Pd 80 Si 20 . When reducing the NG nanopillar diameter to near or double the average grain size, strain softening appears at the larger engineering strain (>20%). Moreover, structural relaxation after a cyclic loading leads to local recovery, and the stress increases upon reloading. We determined Young’s modulus and yield strengths from stress–strain curves of tensile deformations. We find that both properties are smaller in the NG nanopillars as compared with their homogeneous counterparts in both glasses. From Young’s modulus values and the shear band energy, the critical stress for shear band formation is estimated. We find that the predicted critical stress values are quite consistent with the observed deformation modes.
-
![]()
![]()
Why crystalline and glassy lithium thiophosphates are very similar
2020/11/03
A computational study of structure, thermodynamic stability and transport properties
The development of Li+conducting solid electrolytes (SE) is key for the realization of Li all-solid-state batteries. In this context sulfide SE are a promising material class. Using ab-initio molecular dynamics simulations we have studied various LiPS glasses. Stability of these glasses compared to crystals was caclulated as well as their Li-diffusivity. The results reveal that all glasses are thermodynamically meta-stable compared to their crystalline counterparts and that the presence of certain structural units have no influence on transport properties in glasses.
FG Materialmodellierung
Archive
Archive
















Contact
Prof. Dr. rer. nat. Karsten Albe
Head of Research Group

albe@mm.tu-...
fax +49 6151 16-20965
Work
L6|01 211
Postfach 10 06 36
Otto-Berndt-Str. 3
64206
Darmstadt
Gabriele Rühl
Team Assistant

work +49 6151 16-21901
fax +49 6151 16-20965
Work
L6|01 210
Otto-Berndt-Str. 3
64206
Darmstadt