
Allerdings beinhalten solche klassischen Potentiale auch immer einen Kompromiss zwischen Genauigkeit, numerischer Effizienz und Transferabilität. Letztere stellt im Allgemeinen eine große Herausforderung dar: Parametersätze, welche zur Simulation bestimmter Materialeigenschaften oder Prozesse hervorragend geeignet sind, versagen vollständig bei der Vorhersage anderer. Dies ist insbesondere bei der Simulation von amorphen Materialien von erheblicher Bedeutung. Parametersätze, welche aus Eigenschaften kristalliner Referenzstrukturen abgeleitet sind, eignen sich häufig nicht zur Simulation amorpher Strukturen und andersherum. Obwohl diese Problematik weitestgehend bekannt ist, stecken systematische Validierungsansätze die quantitative Analysen der Limitierungen noch immer in ihren Kinderschuhen.
Ziel des vorliegenden Projekts ist es, allgemein anwendbare und robuste Strategien für die Entwicklung interatomarer Potentiale abzuleiten, sowohl kristalline wie auch amorphe Systeme hinreichend genau zu beschreiben. Als Modellsysteme sollen metallische Cu-Zr-(-Al) Legierungen und Gläser sowie keramische Si-O(-C) Verbindungen dienen. Dabei handelt es sich um Systeme, die in der Literatur schon ausgiebig untersucht wurden und für die eine Vielzahl verschiedener Parametrisierungen vorliegt, von der jedoch keine auch nur annähernd die Kriterien einer ausreichenden Transferabilität erfüllt.Zunächst soll versucht werden die Parametrisierungen dieser existierenden, physikalisch motivierten Potentiale zu optimieren. Dies soll durch fitten an ein reichhaltiges Trainings-Set erreicht werden, welches zusätzlich zu kristallinen insbesondere auch amorphe Referenzstrukturen enthält.
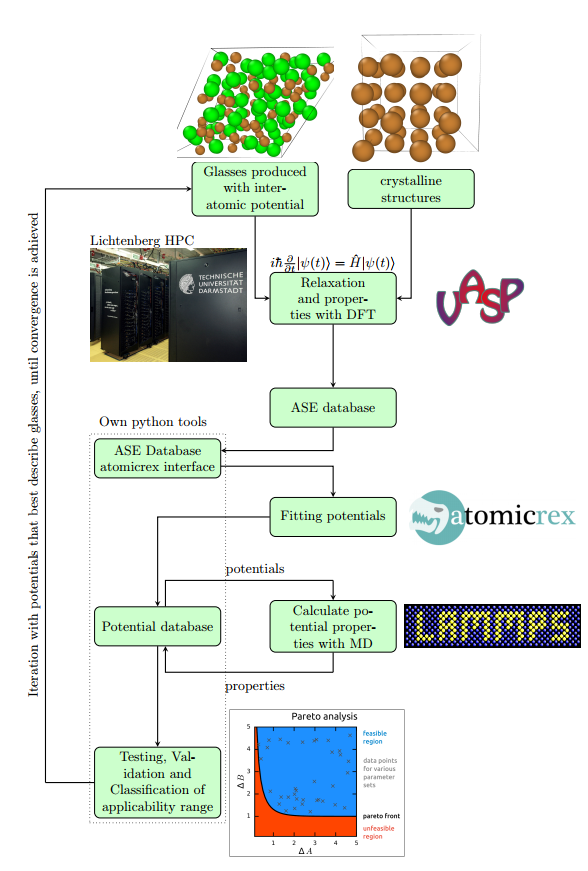
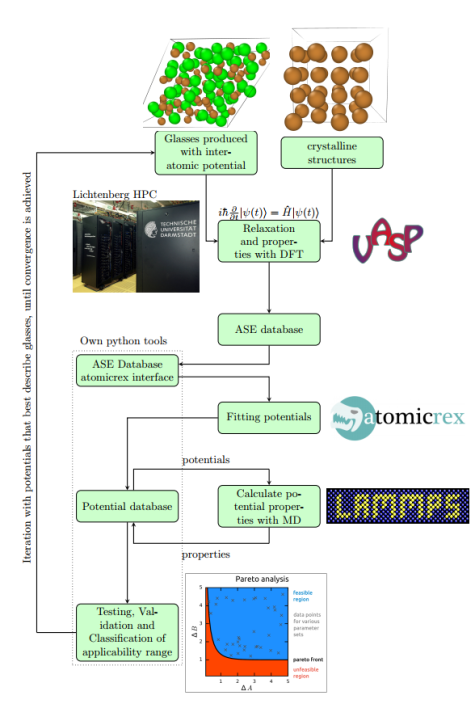
Die Referenzdaten sollen dabei anhand von Dichtefunktionalrechnungen generiert werden. Durch systematische Variation der Gewichtung verschiedener Eigenschaften beim Fitten können anschließend zahlreiche Parametersätze generiert werden. Pareto-Analysen werden dann Aufschluss über die Güte der Potentiale geben und inwiefern eventuelle Transferabilitätsprobleme der Funktionalen Form des Potentials geschuldet sind. Als Alternative bieten sich physikalisch motivierte „second principles“ Potentiale oder neuronale Netzwerkpotentiale an.
Wissenschaftlicher Mitarbeiter

Niklas Leimeroth Dr. rer. nat.
Kontakt
leimeroth@mm.tu-...
work +49 6151 16-21894
Work
L6I01 206
Postfach 10 06 36
Otto-Berndt-Str. 3
64206
Darmstadt
Projektleitung

Jochen Rohrer Ph. D.
Kontakt
rohrer@mm.tu-...
work +49 6151 16-21893
fax +49 6151 16-20965
Work
L6|01 218
Postfach 10 06 36
Otto-Berndt-Str. 3
64206
Darmstadt

