
However, interatomic potentials always represent a compromise between accuracy, numerical efficiency and transferability. Transferability is often challenging: parameter sets that work excellently for one problem setting may fail miserably in another context. This is of particular importance when modelling amorphous materials. Parameter sets that are constructed on the basis of crystalline reference data typically fail for the description of amorphous states and vice versa. Although these transferability issues are well known, systematic validation and testing including a quantitative analysis of the limitations of given parameter sets is still in its infancy.The main objective of this project is to devise robust strategies for the development of interatomic potentials, which are satisfyingly describing both, crystalline and amorphous, states of matter in a thermodynamic consistent manner and can be extended to multi-component systems by using well defined tranining data set optained from first-principles calculations.
We will thereby focus on metallic Cu-Zr-(Al) alloys and glasses and ceramic S-iO(-C) systems. These are well-investigated systems for which a large number of parametrizations exists, all of which, however, have shortcomings in various ways.Initially, we will attempt to improve on existing potentials with physically motivated functional forms. Parameters sets will be obtained by fitting to large training data sets that contain ideal and defective and, most importantly, amorphous reference structures simultaneously. These reference structures will be generated on the basis of ab initio density functional theory calculations. We will systematically study parametrizations obtained by differently weighting various physical properties and characterize the quality and limitations of the potentials in terms of Pareto analysis.
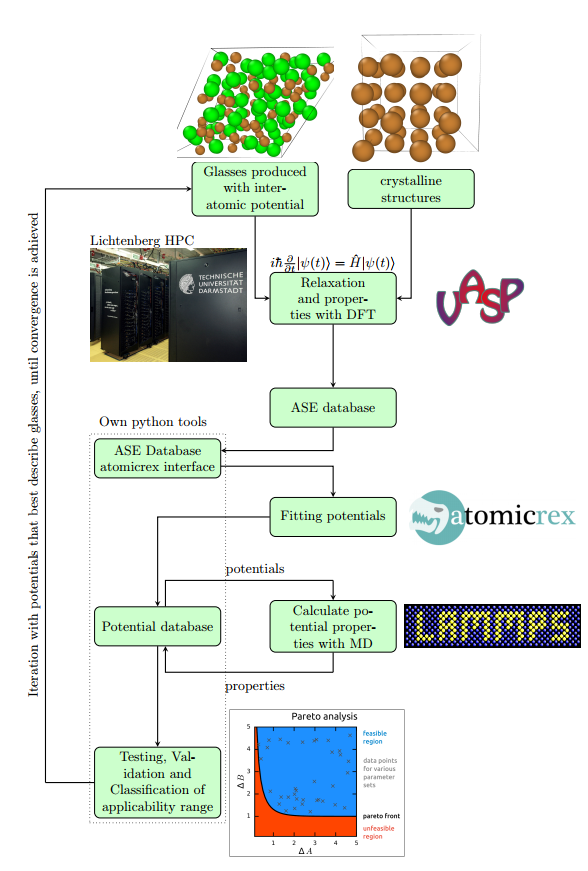
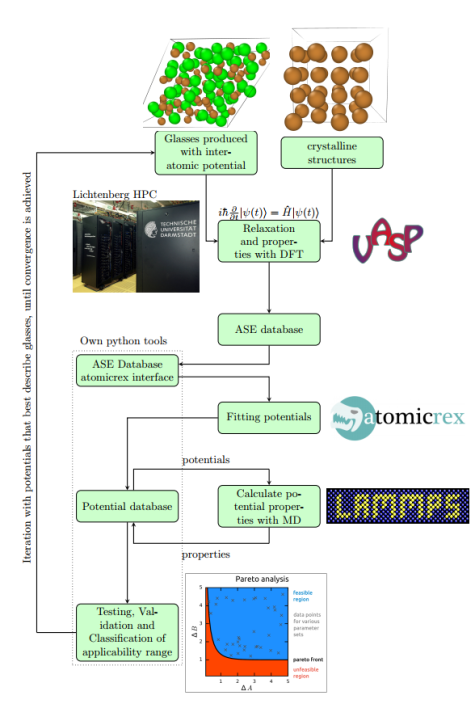
This will allow us to judge on whether a (potentially preserved) limited transferability is due to the functional form of the potentials. As a second strategy will then develop second principles or artificial neural network potentials on the basis of our training sets as these show much higher flexibility than physically motivated functional forms.
Research Assistant

Niklas Leimeroth Dr. rer. nat.
Contact
leimeroth@mm.tu-...
work +49 6151 16-21894
Work
L6I01 206
Otto-Berndt-Str. 3
64206
Darmstadt
Project Leader

Jochen Rohrer Ph. D.
Contact
rohrer@mm.tu-...
work +49 6151 16-21893
fax +49 6151 16-20965
Work
L6|01 218
Otto-Berndt-Str. 3
64206
Darmstadt

